
“An international, interdisciplinary research team of scientists has come up with a machine-learning method that predicts molecular behavior—a breakthrough that can aid in the development of pharmaceuticals.
An international, interdisciplinary research team of scientists has come up with a machine-learning method that predicts molecular behavior, a breakthrough that can aid in the development of pharmaceuticals and the design of new molecules that can be used to enhance the performance of emerging battery technologies, solar cells, and digital displays.
The work appears in the journal Nature Communications.
“By identifying patterns in molecular behavior, the learning algorithm or ‘machine’ we created builds a knowledge base about atomic interactions within a molecule and then draws on that information to predict new phenomena,” explains New York University’s Mark Tuckerman, a professor of chemistry and mathematics and one of the paper’s primary authors.
The paper’s other primary authors were Klaus-Robert Müller of Berlin’s Technische Universität (TUB) and the University of California Irvine’s Kieron Burke.
The work combines innovations in machine learning with physics and chemistry. Data-driven approaches, particularly in the area of machine learning, allow everyday devices to learn automatically from limited sample data and, subsequently, to act on new input information. Such approaches have transformed how we carry out common tasks like online searching, text analysis, image recognition, and language translation.
In recent years, related development has occurred in the natural sciences, with efforts directed toward engineering, materials science, and molecular design. However, machine- learning approaches in these fields have generally not explored the creation of methodologies—tools that could advance science in ways that have already been achieved in banking and public safety.
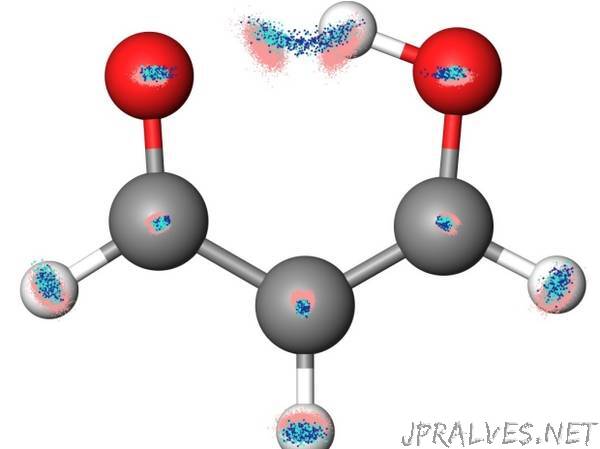
The research team created a machine that can learn complex interatomic interactions, which are normally prescribed by complex quantum mechanical calculations, without having to perform such intricate calculations.
In constructing their machine, the researchers created a small sample set of the molecule they wished to study in order to train the algorithm and then used the machine to simulate complex chemical behavior within the molecule. As an illustrative example, they chose a chemical process that occurs within a simple molecule known as malonaldehyde. To weigh the viability of the tool, they examined how the machine predicted the chemical behavior and then compared their prediction with our current chemical understanding of the molecule. The results revealed how much the machine could learn from the limited training data it had been given.
“Now we have reached the ability to not only use AI to learn from data, but we can probe the AI model to further our scientific understanding and gain new insights,” remarks Klaus-Robert Müller, professor for machine learning at Technical University of Berlin.
A video demonstrating, for the first time, a chemical process that was modelled by machine learning — a proton transferring within the malonaldehyde molecule — can be viewed here: http://bit.ly/2xaTuPn.
The paper’s other authors also include Felix Brockherde, the lead author, who is a Ph.D. student in computer science and software engineering at the TUB, Leslie Vogt, a postdoctoral researcher in chemistry at NYU, and Li Li, a recent graduate from UC Irvine.
The study was supported, in part, by grants from the U.S. Army Research Office (W911NF-13-1-0387), the U.S. National Science Foundation (CHE 1464795), the Information & Communications Technology Promotion (IITP) of the Korean government (No. 2017-0-00451), and the Einstein Foundation.”