
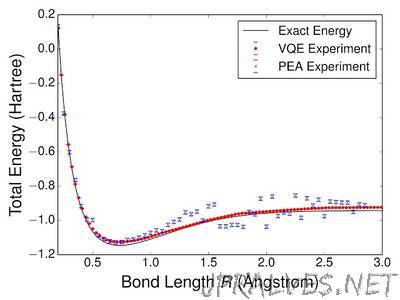
“One of the most promising applications of quantum computing is the ability to efficiently model quantum systems in nature that are considered intractable for classical computers. Now, in collaboration with the Aspuru-Guzik group at Harvard and researchers from Lawrence Berkeley National Labs, UC Santa Barbara, Tufts University and University College London, we have performed the first completely scalable quantum simulation of a molecule. Our experimental results are detailed in the paper Scalable Quantum Simulation of Molecular Energies, which recently appeared in Physical Review X. The goal of our experiment was to use quantum hardware to efficiently solve the molecular electronic structure problem, which seeks the solution for the lowest energy configuration of electrons in the presence of a given nuclear configuration. In order to predict chemical reaction rates (which govern the mechanism of chemical reactions), one must make these calculations to extremely high precision. The ability to predict such rates could revolutionize the design of solar cells, industrial catalysts, batteries, flexible electronics, medicines, materials and more. The primary difficulty is that molecular systems form highly entangled quantum superposition states which require exponentially many classical computing resources in order to represent to sufficiently high precision. For example, exactly computing the energies of methane (CH4) takes about one second, but the same calculation takes about ten minutes for ethane (C2H6) and about ten days for propane (C3H8).”