
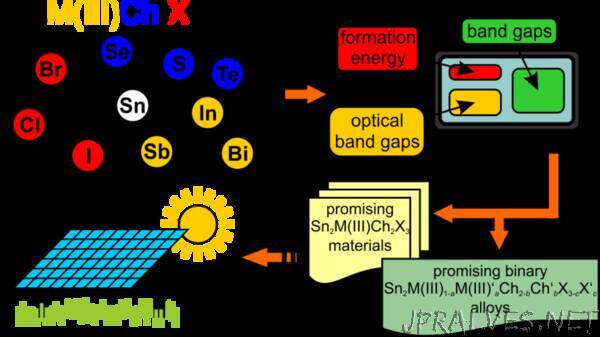
“CEST researchers have successfully identified new and promising candidates for photovoltaic applications using computational methods.
The CEST (Computational Electronic Structure Theory) group members Pascal Henkel and Patrick Rinke, in collaboration with researchers from the University of Tampere (Finland) and Xi’an University (China), have recently published a study in which they explore properties of mixed-metal chalcohalides using Density Functional Theory (DFT). Mixed-metal Chalcohalides are a class of compounds with potential interest for photovoltaic applications.
These semiconducting materials combine the optoelectronic properties of halide perovskites with the stability of metal chalcogenides. As a result, lead-free mixed-metal chalcohalides have the potential to overcome the stability and toxicity problems of other known halide perovskites and to facilitate the commercialization of novel solar cells. The objective of this study was to identify new mixed-metal chalcohalide materials and alloys that are stable and also suitable for indoor and/or outdoor photovoltaic applications. For this purpose, a total of 27 mixed-metal chalcohalides compounds were considered. In total, 12 promising materials were identified. Nine of these compounds are so far unknown and have been investigated for the first time in this study. In addition, the researchers identified 12 mixed-metal chalcohalides alloys that are suitable for photovoltaic applications and that offer the possibility to fine tune the materials properties for specific lighting conditions. The study provides a first step towards further research on mixed-metal chalcohalides and their future devices.
So far, mixed-metal chalcohalide compounds have been scarcely explored and only a few of the compounds in the study were previously known (or had even been synthesized). Materials discovery of new mixed-metal chalcohalides through experimental synthesis is generally very time-consuming. Computational tools such as DFT explore materials in silico and can lead the way with accelerated materials discovery. The article was published in Chemistry of Materials.”